Overview
BANKSY is a method for clustering spatial omics data by augmenting the features of each cell with both an average of the features of its spatial neighbors along with neighborhood feature gradients. By incorporating neighborhood information for clustering, BANKSY is able to
- improve cell-type assignment in noisy data
- distinguish subtly different cell-types stratified by microenvironment
- identify spatial domains sharing the same microenvironment
BANKSY is applicable to a wide array of spatial technologies (e.g. 10x Visium, Slide-seq, MERFISH, CosMX, CODEX) and scales well to large datasets. For more details, check out:
- the paper,
- the peer review file,
- a tweetorial on BANKSY,
- a set of vignettes showing basic usage,
- usage compatibility with Seurat (here and here),
- a Python version of this package,
- a Zenodo archive containing scripts to reproduce the analyses in the paper, and the corresponding GitHub Pages (and here for analyses done in Python).
Installation
The Banksy package can be installed via Bioconductor. This currently requires R >= 4.4.0.
BiocManager::install('Banksy')To install directly from GitHub instead, use
remotes::install_github("prabhakarlab/Banksy")To use the legacy version of Banksy utilising the BanksyObject class, use
remotes::install_github("prabhakarlab/Banksy@legacy")Banksy is also interoperable with Seurat via SeuratWrappers. Documentation on how to run BANKSY on Seurat objects can be found here. For installation of SeuratWrappers with BANKSY version >= 0.1.6, run
remotes::install_github('satijalab/seurat-wrappers')Quick start
Load BANKSY. We’ll also load SpatialExperiment and SummarizedExperiment for containing and manipulating the data, scuttle for normalization and quality control, and scater, ggplot2 and cowplot for visualisation.
library(Banksy)
library(SummarizedExperiment)
library(SpatialExperiment)
library(scuttle)
library(scater)
library(cowplot)
library(ggplot2)Here, we’ll run BANKSY on mouse hippocampus data.
Initialize a SpatialExperiment object and perform basic quality control and normalization.
se <- SpatialExperiment(assay = list(counts = gcm), spatialCoords = locs)
# QC based on total counts
qcstats <- perCellQCMetrics(se)
thres <- quantile(qcstats$total, c(0.05, 0.98))
keep <- (qcstats$total > thres[1]) & (qcstats$total < thres[2])
se <- se[, keep]
# Normalization to mean library size
se <- computeLibraryFactors(se)
aname <- "normcounts"
assay(se, aname) <- normalizeCounts(se, log = FALSE)Compute the neighborhood matrices for BANKSY. Setting compute_agf=TRUE computes both the weighted neighborhood mean () and the azimuthal Gabor filter (). The number of spatial neighbors used to compute and are k_geom[1]=15 and k_geom[2]=30 respectively. We run BANKSY at lambda=0 corresponding to non-spatial clustering, and lambda=0.2 corresponding to BANKSY for cell-typing.
An important note about choosing the
lambdaparameter for the older Visium v1 / v2 55um datasets or the original ST 100um technology:For most modern high resolution technologies like Xenium, Visium HD, StereoSeq, MERFISH, STARmap PLUS, SeqFISH+, SlideSeq v2, and CosMx (and others), we recommend the usual defults for
lambda: For cell typing, uselambda = 0.2(as shown below, or in this vignette) and for domain segmentation, uselambda = 0.8. These technologies are either imaging based, having true single-cell resolution (e.g., MERFISH), or are sequencing based, having barcoded spots on the scale of single-cells (e.g., Visium HD). We find that the usual defaults work well at this measurement resolution.However, for the older Visium v1/v2 or ST technologies, with their much lower resolution spots (55um and 100um diameter, respectively), we find that
lambda = 0.2seems to work best for domain segmentation. This could be because each spot already measures the average transcriptome of several cells in a neighbourhood. It seems thatlambda = 0.2shares enough information between these neighbourhoods to lead to good domain segmentation performance. For example, in the human DLPFC vignette, we uselambda = 0.2on a Visium v1/v2 dataset. Also note that in these lower resolution technologies, each spot can have multiple cells of different types, and as such cell-typing is not defined for them.
lambda <- c(0, 0.2)
k_geom <- c(15, 30)
se <- Banksy::computeBanksy(se, assay_name = aname, compute_agf = TRUE, k_geom = k_geom)
#> Computing neighbors...
#> Spatial mode is kNN_median
#> Parameters: k_geom=15
#> Done
#> Computing neighbors...
#> Spatial mode is kNN_median
#> Parameters: k_geom=30
#> Done
#> Computing harmonic m = 0
#> Using 15 neighbors
#> Done
#> Computing harmonic m = 1
#> Using 30 neighbors
#> Centering
#> DoneNext, run PCA on the BANKSY matrix and perform clustering. Setting use_agf=TRUE uses both and to construct the BANKSY matrix.
set.seed(1000)
se <- Banksy::runBanksyPCA(se, use_agf = TRUE, lambda = lambda)
se <- Banksy::runBanksyUMAP(se, use_agf = TRUE, lambda = lambda)
se <- Banksy::clusterBanksy(se, use_agf = TRUE, lambda = lambda, resolution = 1.2)Different clustering runs can be relabeled to minimise their differences with connectClusters:
se <- Banksy::connectClusters(se)
#> clust_M1_lam0.2_k50_res1.2 --> clust_M1_lam0_k50_res1.2Visualise the clustering output for non-spatial clustering (lambda=0) and BANKSY clustering (lambda=0.2).
cnames <- colnames(colData(se))
cnames <- cnames[grep("^clust", cnames)]
colData(se) <- cbind(colData(se), spatialCoords(se))
plot_nsp <- plotColData(se,
x = "sdimx", y = "sdimy",
point_size = 0.6, colour_by = cnames[1]
)
plot_bank <- plotColData(se,
x = "sdimx", y = "sdimy",
point_size = 0.6, colour_by = cnames[2]
)
plot_grid(plot_nsp + coord_equal(), plot_bank + coord_equal(), ncol = 2)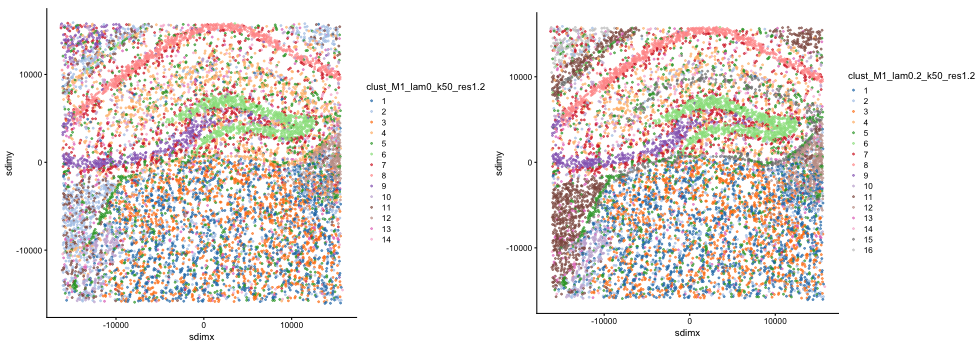
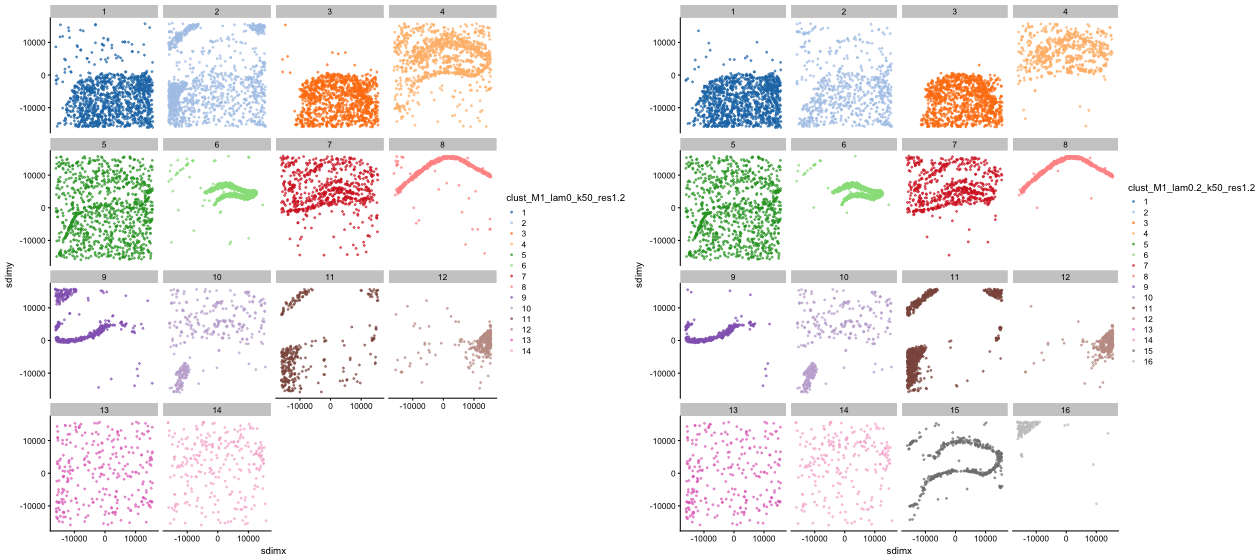
For clarity, we can visualise each of the clusters separately:
plot_grid(
plot_nsp + facet_wrap(~colour_by),
plot_bank + facet_wrap(~colour_by),
ncol = 2
)
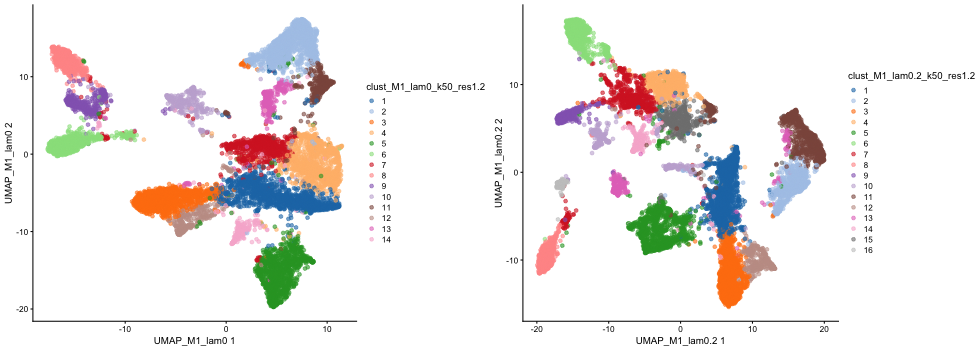
Visualize UMAPs of the non-spatial and BANKSY embedding:
rdnames <- reducedDimNames(se)
umap_nsp <- plotReducedDim(se,
dimred = grep("UMAP.*lam0$", rdnames, value = TRUE),
colour_by = cnames[1]
)
umap_bank <- plotReducedDim(se,
dimred = grep("UMAP.*lam0.2$", rdnames, value = TRUE),
colour_by = cnames[2]
)
plot_grid(
umap_nsp,
umap_bank,
ncol = 2
)
Session information
sessionInfo()
#> R version 4.3.2 (2023-10-31)
#> Platform: aarch64-apple-darwin20 (64-bit)
#> Running under: macOS Sonoma 14.2.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.3-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.11.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: America/Detroit
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] cowplot_1.1.3 scater_1.30.1
#> [3] ggplot2_3.4.4 scuttle_1.12.0
#> [5] SpatialExperiment_1.12.0 SingleCellExperiment_1.24.0
#> [7] SummarizedExperiment_1.32.0 Biobase_2.62.0
#> [9] GenomicRanges_1.54.1 GenomeInfoDb_1.38.6
#> [11] IRanges_2.36.0 S4Vectors_0.40.2
#> [13] BiocGenerics_0.48.1 MatrixGenerics_1.14.0
#> [15] matrixStats_1.2.0 Banksy_0.99.12
#>
#> loaded via a namespace (and not attached):
#> [1] tidyselect_1.2.0 viridisLite_0.4.2
#> [3] farver_2.1.1 dplyr_1.1.4
#> [5] vipor_0.4.7 viridis_0.6.5
#> [7] bitops_1.0-7 fastmap_1.1.1
#> [9] RCurl_1.98-1.14 digest_0.6.34
#> [11] rsvd_1.0.5 lifecycle_1.0.4
#> [13] magrittr_2.0.3 dbscan_1.1-12
#> [15] compiler_4.3.2 rlang_1.1.3
#> [17] tools_4.3.2 igraph_2.0.1.1
#> [19] utf8_1.2.4 yaml_2.3.8
#> [21] data.table_1.15.0 knitr_1.45
#> [23] labeling_0.4.3 S4Arrays_1.2.0
#> [25] mclust_6.0.1 DelayedArray_0.28.0
#> [27] abind_1.4-5 BiocParallel_1.36.0
#> [29] withr_3.0.0 grid_4.3.2
#> [31] fansi_1.0.6 beachmat_2.18.0
#> [33] colorspace_2.1-0 aricode_1.0.3
#> [35] scales_1.3.0 cli_3.6.2
#> [37] rmarkdown_2.25 crayon_1.5.2
#> [39] leidenAlg_1.1.2 generics_0.1.3
#> [41] rstudioapi_0.15.0 rjson_0.2.21
#> [43] DelayedMatrixStats_1.24.0 ggbeeswarm_0.7.2
#> [45] RcppHungarian_0.3 zlibbioc_1.48.0
#> [47] parallel_4.3.2 XVector_0.42.0
#> [49] vctrs_0.6.5 Matrix_1.6-5
#> [51] BiocSingular_1.18.0 BiocNeighbors_1.20.2
#> [53] ggrepel_0.9.5 irlba_2.3.5.1
#> [55] beeswarm_0.4.0 magick_2.8.2
#> [57] glue_1.7.0 codetools_0.2-19
#> [59] uwot_0.1.16 RcppAnnoy_0.0.22
#> [61] gtable_0.3.4 ScaledMatrix_1.10.0
#> [63] munsell_0.5.0 tibble_3.2.1
#> [65] pillar_1.9.0 htmltools_0.5.7
#> [67] GenomeInfoDbData_1.2.11 R6_2.5.1
#> [69] sparseMatrixStats_1.14.0 evaluate_0.23
#> [71] sccore_1.0.4 lattice_0.22-5
#> [73] highr_0.10 Rcpp_1.0.12
#> [75] gridExtra_2.3 SparseArray_1.2.4
#> [77] xfun_0.42 pkgconfig_2.0.3