Domain segmentation (STARmap PLUS mouse brain)
Source:vignettes/domain-segment.Rmd
domain-segment.RmdHere, we demonstrate BANKSY domain segmentation on a STARmap PLUS dataset of the mouse brain from Shi et al. (2022).
library(Banksy)
library(data.table)
library(SummarizedExperiment)
library(SpatialExperiment)
library(scater)
library(cowplot)
library(ggplot2)Data preprocessing
Data from the study is available from the Single
Cell Portal. We analyze data from well11. The data
comprise 1,022 genes profiled at subcellular resolution in 43,341
cells.
#' Change paths accordingly
gcm_path <- "../data/well11processed_expression_pd.csv.gz"
mdata_path <- "../data/well11_spatial.csv.gz"
#' Gene cell matrix
gcm <- fread(gcm_path)
genes <- gcm$GENE
gcm <- as.matrix(gcm[, -1])
rownames(gcm) <- genes
#' Spatial coordinates and metadata
mdata <- fread(mdata_path, skip = 1)
headers <- names(fread(mdata_path, nrows = 0))
colnames(mdata) <- headers
#' Orient spatial coordinates
xx <- mdata$X
yy <- mdata$Y
mdata$X <- max(yy) - yy
mdata$Y <- max(xx) - xx
mdata <- data.frame(mdata)
rownames(mdata) <- colnames(gcm)
locs <- as.matrix(mdata[, c("X", "Y", "Z")])
#' Create SpatialExperiment
se <- SpatialExperiment(
assay = list(processedExp = gcm),
spatialCoords = locs,
colData = mdata
)Running BANKSY
Run BANKSY in domain segmentation mode with lambda=0.8.
This places larger weights on the mean neighborhood expression and
azimuthal Gabor filter in constructing the BANKSY matrix. We adjust the
resolution to yield 23 clusters based on the results from Maher et
al. (2023) (see Fig. 1, 2).
Note that the parameter values for domain segmentation for datasets generated using the older Visium v1 / v2 55um technologies are
lambda = 0.2andk_geom = 18. See the note in the tutorial on the main page for more details.
lambda <- 0.8
k_geom <- 30
npcs <- 50
aname <- "processedExp"
se <- Banksy::computeBanksy(se, assay_name = aname, k_geom = k_geom)
set.seed(1000)
se <- Banksy::runBanksyPCA(se, lambda = lambda, npcs = npcs)
set.seed(1000)
se <- Banksy::clusterBanksy(se, lambda = lambda, npcs = npcs, resolution = 0.8)Cluster labels are stored in the colData slot:
head(colData(se))
#> DataFrame with 6 rows and 4 columns
#> X Y clust_M1_lam0.8_k50_res0.8 sample_id
#> <numeric> <numeric> <factor> <character>
#> 1 24225.5 23984.2 10 sample01
#> 2 24849.2 22679.1 10 sample01
#> 3 24488.3 22970.3 10 sample01
#> 4 24371.4 23727.5 10 sample01
#> 5 24362.2 23300.6 10 sample01
#> 6 24644.5 23112.8 10 sample01Visualize clustering results:
cnames <- colnames(colData(se))
cnames <- cnames[grep("^clust", cnames)]
plotColData(se, x = "X", y = "Y", point_size = 0.01, colour_by = cnames[1]) +
scale_color_manual(values = pals::glasbey()) +
coord_equal() +
theme(legend.position = "none")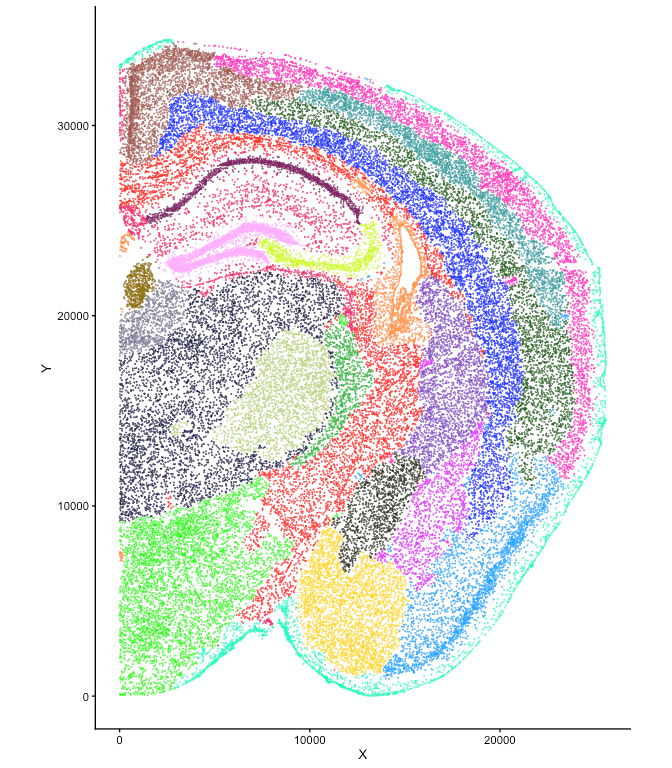
Session information
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: aarch64-apple-darwin20
#> Running under: macOS Sonoma 14.6.1
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> time zone: Europe/London
#> tzcode source: internal
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] cowplot_1.1.3 scater_1.32.1
#> [3] ggplot2_3.5.1 scuttle_1.14.0
#> [5] SpatialExperiment_1.14.0 SingleCellExperiment_1.26.0
#> [7] SummarizedExperiment_1.34.0 Biobase_2.64.0
#> [9] GenomicRanges_1.56.1 GenomeInfoDb_1.40.1
#> [11] IRanges_2.38.1 S4Vectors_0.42.1
#> [13] BiocGenerics_0.48.1 MatrixGenerics_1.16.0
#> [15] matrixStats_1.3.0 data.table_1.15.4
#> [17] Banksy_1.5.0 BiocStyle_2.32.1
#>
#> loaded via a namespace (and not attached):
#> [1] gridExtra_2.3 rlang_1.1.4
#> [3] magrittr_2.0.3 compiler_4.4.1
#> [5] sccore_1.0.5 DelayedMatrixStats_1.26.0
#> [7] systemfonts_1.1.0 vctrs_0.6.5
#> [9] pkgconfig_2.0.3 crayon_1.5.3
#> [11] fastmap_1.2.0 magick_2.8.4
#> [13] XVector_0.44.0 utf8_1.2.4
#> [15] rmarkdown_2.27 UCSC.utils_1.0.0
#> [17] ggbeeswarm_0.7.2 ragg_1.3.2
#> [19] xfun_0.52 zlibbioc_1.50.0
#> [21] cachem_1.1.0 beachmat_2.20.0
#> [23] jsonlite_1.8.8 DelayedArray_0.30.1
#> [25] BiocParallel_1.38.0 irlba_2.3.5.1
#> [27] parallel_4.4.1 aricode_1.0.3
#> [29] R6_2.5.1 bslib_0.8.0
#> [31] leidenAlg_1.1.3 jquerylib_0.1.4
#> [33] Rcpp_1.0.13 bookdown_0.43
#> [35] knitr_1.48 Matrix_1.7-0
#> [37] igraph_2.0.3 tidyselect_1.2.1
#> [39] rstudioapi_0.16.0 abind_1.4-5
#> [41] yaml_2.3.10 viridis_0.6.5
#> [43] codetools_0.2-20 lattice_0.22-6
#> [45] tibble_3.2.1 withr_3.0.1
#> [47] evaluate_0.24.0 desc_1.4.3
#> [49] mclust_6.1.1 pillar_1.9.0
#> [51] BiocManager_1.30.23 generics_0.1.3
#> [53] dbscan_1.2-0 sparseMatrixStats_1.16.0
#> [55] munsell_0.5.1 scales_1.3.0
#> [57] glue_1.7.0 tools_4.4.1
#> [59] BiocNeighbors_1.22.0 ScaledMatrix_1.12.0
#> [61] fs_1.6.4 grid_4.4.1
#> [63] colorspace_2.1-1 GenomeInfoDbData_1.2.11
#> [65] RcppHungarian_0.3 beeswarm_0.4.0
#> [67] BiocSingular_1.20.0 vipor_0.4.7
#> [69] cli_3.6.3 rsvd_1.0.5
#> [71] textshaping_0.4.0 fansi_1.0.6
#> [73] viridisLite_0.4.2 S4Arrays_1.4.1
#> [75] dplyr_1.1.4 uwot_0.2.2
#> [77] gtable_0.3.5 sass_0.4.9
#> [79] digest_0.6.36 SparseArray_1.4.8
#> [81] ggrepel_0.9.5 rjson_0.2.21
#> [83] htmlwidgets_1.6.4 htmltools_0.5.8.1
#> [85] pkgdown_2.1.0 lifecycle_1.0.4
#> [87] httr_1.4.7